
375240
HNF4 Antagonist, BI6015
The HNF4 Antagonist, BI6, also referenced under CAS 93987-29-2, controls the biological activity of HNF4.
別名:
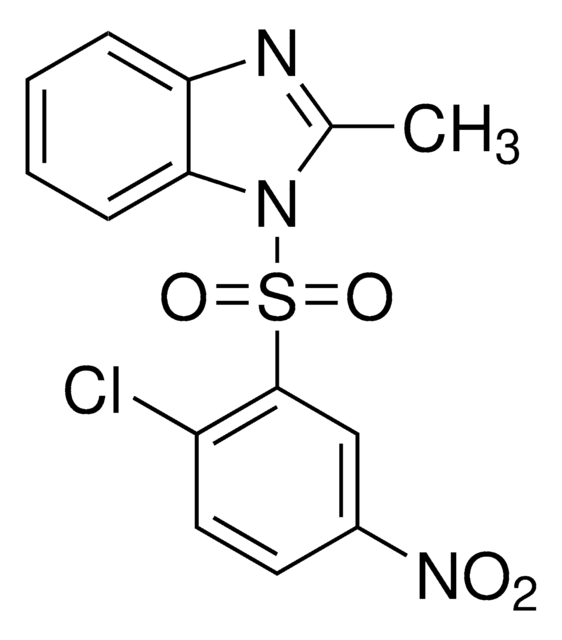
HNF4 Antagonist, BI6015, 2-Methyl-1-(2-methyl-5-nitrophenylsulfonyl)-1H-benzo[d]imidazole, Hepatocyte Nuclear Factor4 Antagonist
About This Item
おすすめの製品



品質水準
アッセイ
≥99% (HPLC)
フォーム
powder
メーカー/製品名
Calbiochem®
保管条件
OK to freeze
protect from light
色
beige
溶解性
DMSO: 10 mg/mL
輸送温度
ambient
保管温度
2-8°C
SMILES記法
CC1=NC2=C(N1S(C3=CC([N+]([O-])=O)=CC=C3C)(=O)=O)C=CC=C2
InChI
1S/C15H13N3O4S/c1-10-7-8-12(18(19)20)9-15(10)23(21,22)17-11(2)16-13-5-3-4-6-14(13)17/h3-9H,1-2H3
InChI Key
ILVCPQPMRPHZSG-UHFFFAOYSA-N
詳細
生物化学的/生理学的作用
HNFα & γ
包装
警告
その他情報
法的情報
保管分類コード
11 - Combustible Solids
WGK
WGK 2
引火点(°F)
Not applicable
引火点(℃)
Not applicable
適用法令
試験研究用途を考慮した関連法令を主に挙げております。化学物質以外については、一部の情報のみ提供しています。 製品を安全かつ合法的に使用することは、使用者の義務です。最新情報により修正される場合があります。WEBの反映には時間を要することがあるため、適宜SDSをご参照ください。
Jan Code
375240-MG:
375240-25MG:
試験成績書(COA)
製品のロット番号・バッチ番号を入力して、試験成績書(COA) を検索できます。ロット番号・バッチ番号は、製品ラベルに「Lot」または「Batch」に続いて記載されています。
ライフサイエンス、有機合成、材料科学、クロマトグラフィー、分析など、あらゆる分野の研究に経験のあるメンバーがおります。.
製品に関するお問い合わせはこちら(テクニカルサービス)