
43659
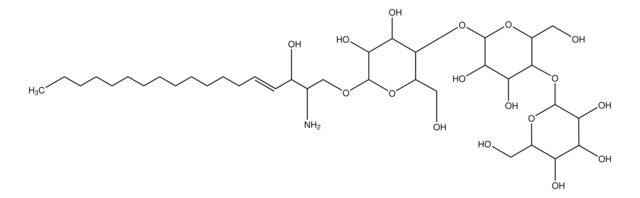
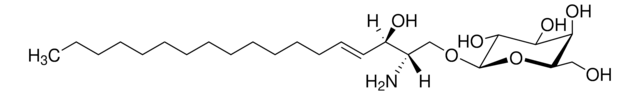
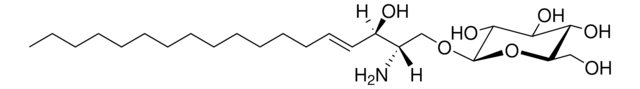
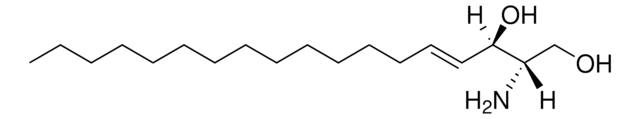
Glucosylsphingosine
≥98.0% (TLC)
Sinônimo(s):
(2S,3R,4E)-2-Amino-3-hydroxy-4-octadecen-1-yl β-D-glucopyranoside, 1-β-D-Glucosylsphingosine, Glucosyl-C18-sphingosine
About This Item
Produtos recomendados
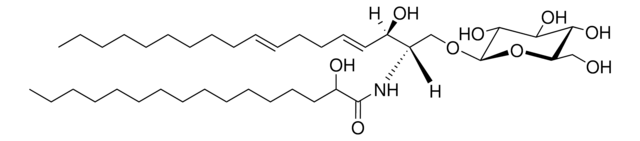

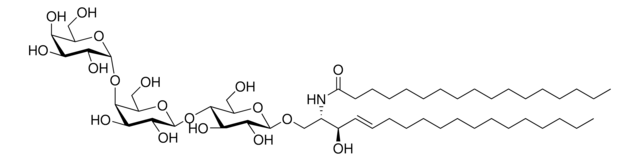

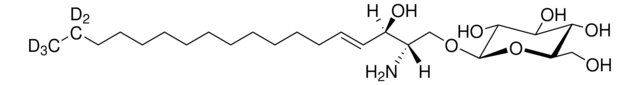
Nível de qualidade
Ensaio
≥98.0% (TLC)
Formulário
powder
tipo de lipídio
sphingolipids
temperatura de armazenamento
−20°C
cadeia de caracteres SMILES
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
chave InChI
HHJTWTPUPVQKNA-JLRUQHRASA-N
Ações bioquímicas/fisiológicas
Código de classe de armazenamento
11 - Combustible Solids
Classe de risco de água (WGK)
WGK 3
Ponto de fulgor (°F)
Not applicable
Ponto de fulgor (°C)
Not applicable
Escolha uma das versões mais recentes:
Já possui este produto?
Encontre a documentação dos produtos que você adquiriu recentemente na biblioteca de documentos.
Os clientes também visualizaram
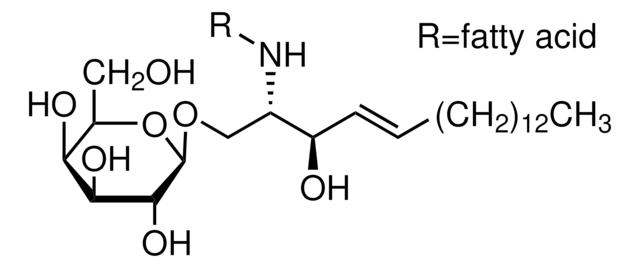

Nossa equipe de cientistas tem experiência em todas as áreas de pesquisa, incluindo Life Sciences, ciência de materiais, síntese química, cromatografia, química analítica e muitas outras.
Entre em contato com a assistência técnica