
05-583
Przeciwciało anty-CFTR, klon M3A7
clone M3A7, Upstate®, from mouse
Synonim(y):
ATP-binding cassette sub-family C, member 7, ATP-binding cassette transporter sub-family C member 7, cAMP-dependent chloride channel, cystic fibrosis transmembrane conductance regulator, cystic fibrosis transmembrane conductance regulator (ATP-binding ca
About This Item
Polecane produkty





pochodzenie biologiczne
mouse
Poziom jakości
forma przeciwciała
purified immunoglobulin
rodzaj przeciwciała
primary antibodies
klon
M3A7, monoclonal
reaktywność gatunkowa
human
producent / nazwa handlowa
Upstate®
metody
immunohistochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
izotyp
IgG1
numer dostępu NCBI
numer dostępu UniProt
Warunki transportu
dry ice
docelowa modyfikacja potranslacyjna
unmodified
informacje o genach
human ... CFTR(1080)
Opis ogólny
Specyficzność
Immunogen
Zastosowanie
To przeciwciało zostało zgłoszone do immunoprecypitacji CFTR. (Kartner, N., 1998.)
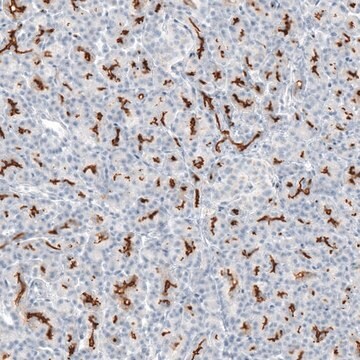
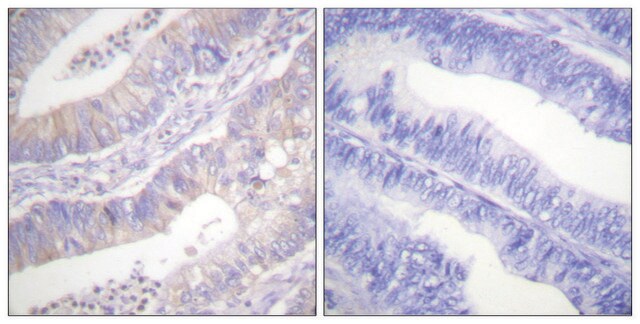
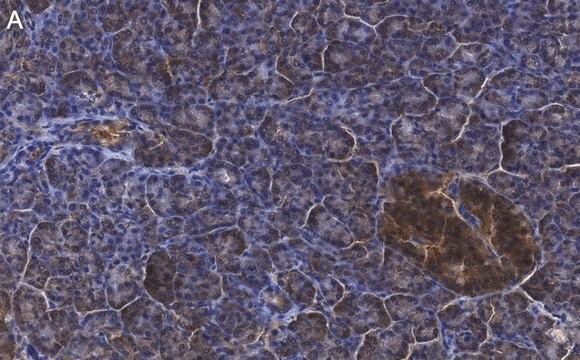
Immunohistochemia:
To przeciwciało zostało zgłoszone do immunobarwienia CFTR w skrawkach tkanki ludzkiej trzustki. (Kartner, N., 1998.)
Neuroscience
Kanały jonowe i transportery
Jakość
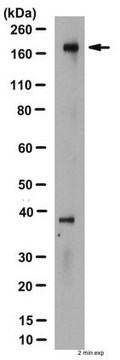
Western Blot Analysis:
0.5-2 µg/mL of this lot detected CFTR from 20-50 µg of human T84 colon carcinoma epithelial RIPA cell lysates. 0.5-2 µg/mL of a previous lot detected CFTR from CFTR-transfected BHK (Haardt, M., 1999).
Note: Do not boil the lysate. Instead incubate at 37°C for 30 minutes. CFTR can run as a diffuse protein on SDS-PAGE.
Opis wartości docelowych
Postać fizyczna
Przechowywanie i stabilność
Zalecenia dotyczące postępowania:
Po otrzymaniu, a przed zdjęciem nasadki, odwirować fiolkę i delikatnie wymieszać roztwór. Rozdzielić do probówek mikrowirówkowych i przechowywać w temperaturze -20°C. Unikać powtarzających się cykli zamrażania/rozmrażania, które mogą uszkodzić IgG i wpłynąć na działanie produktu. Uwaga: Zmienność temperatur w zamrażarce poniżej -20°C może powodować zamarzanie roztworów zawierających glicerol podczas przechowywania.
Komentarz do analizy
Lizat komórek T84.
Inne uwagi
Informacje prawne
Oświadczenie o zrzeczeniu się odpowiedzialności
Nie możesz znaleźć właściwego produktu?
Wypróbuj nasz Narzędzie selektora produktów.
polecane
Kod klasy składowania
10 - Combustible liquids
Klasa zagrożenia wodnego (WGK)
WGK 1
Certyfikaty analizy (CoA)
Poszukaj Certyfikaty analizy (CoA), wpisując numer partii/serii produktów. Numery serii i partii można znaleźć na etykiecie produktu po słowach „seria” lub „partia”.
Masz już ten produkt?
Dokumenty związane z niedawno zakupionymi produktami zostały zamieszczone w Bibliotece dokumentów.
Nasz zespół naukowców ma doświadczenie we wszystkich obszarach badań, w tym w naukach przyrodniczych, materiałoznawstwie, syntezie chemicznej, chromatografii, analityce i wielu innych dziedzinach.
Skontaktuj się z zespołem ds. pomocy technicznej