
05-583
Anti-CFTR Antibody, clone M3A7
clone M3A7, Upstate®, from mouse
Sinônimo(s):
ATP-binding cassette sub-family C, member 7, ATP-binding cassette transporter sub-family C member 7, cAMP-dependent chloride channel, cystic fibrosis transmembrane conductance regulator, cystic fibrosis transmembrane conductance regulator (ATP-binding ca
About This Item
Produtos recomendados




fonte biológica
mouse
Nível de qualidade
forma do anticorpo
purified immunoglobulin
tipo de produto de anticorpo
primary antibodies
clone
M3A7, monoclonal
reatividade de espécies
human
fabricante/nome comercial
Upstate®
técnica(s)
immunohistochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
Isotipo
IgG1
nº de adesão NCBI
nº de adesão UniProt
Condições de expedição
dry ice
modificação pós-traducional do alvo
unmodified
Informações sobre genes
human ... CFTR(1080)
Descrição geral
Especificidade
Imunogênio
Aplicação
This antibody has been reported to immunoprecipitate CFTR. (Kartner, N., 1998.)
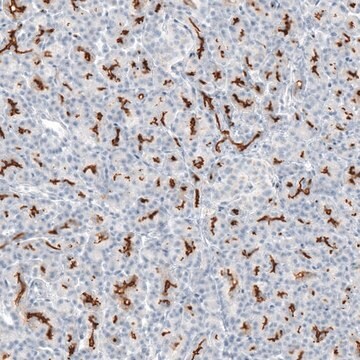
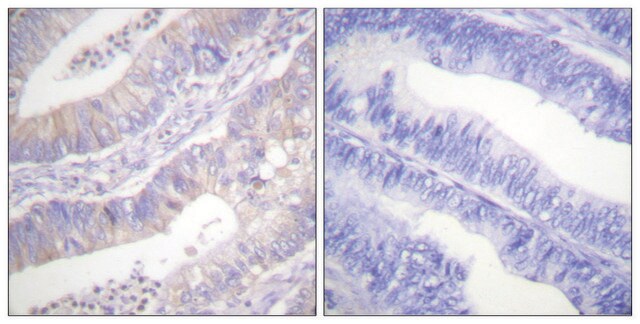
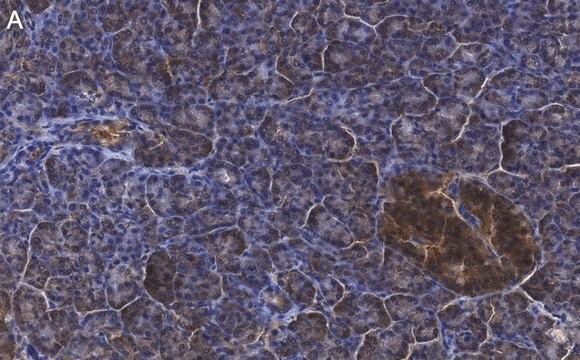
Immunohistochemistry:
This antibody has been reported to immunostain CFTR in human pancreatic tissue sections. (Kartner, N., 1998.)
Neuroscience
Ion Channels & Transporters
Qualidade
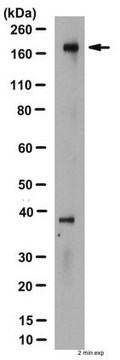
Western Blot Analysis:
0.5-2 µg/mL of this lot detected CFTR from 20-50 µg of human T84 colon carcinoma epithelial RIPA cell lysates. 0.5-2 µg/mL of a previous lot detected CFTR from CFTR-transfected BHK (Haardt, M., 1999).
Note: Do not boil the lysate. Instead incubate at 37°C for 30 minutes. CFTR can run as a diffuse protein on SDS-PAGE.
Descrição-alvo
forma física
Armazenamento e estabilidade
Handling Recommendations:
Upon receipt, and prior to removing the cap, centrifuge the vial and gently mix the solution. Aliquot into microcentrifuge tubes and store at -20°C. Avoid repeated freeze/thaw cycles, which may damage IgG and affect product performance. Note: Variability in freezer temperatures below -20°C may cause glycerol-containing solutions to become frozen during storage.
Nota de análise
T84 cell lysate.
Outras notas
Informações legais
Exoneração de responsabilidade
Não está encontrando o produto certo?
Experimente o nosso Ferramenta de seleção de produtos.
recomendado
Código de classe de armazenamento
10 - Combustible liquids
Classe de risco de água (WGK)
WGK 1
Certificados de análise (COA)
Busque Certificados de análise (COA) digitando o Número do Lote do produto. Os números de lote e remessa podem ser encontrados no rótulo de um produto após a palavra “Lot” ou “Batch”.
Já possui este produto?
Encontre a documentação dos produtos que você adquiriu recentemente na biblioteca de documentos.
Nossa equipe de cientistas tem experiência em todas as áreas de pesquisa, incluindo Life Sciences, ciência de materiais, síntese química, cromatografia, química analítica e muitas outras.
Entre em contato com a assistência técnica