
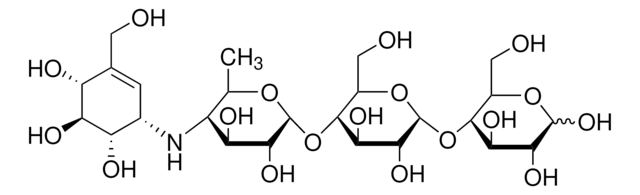
B8299
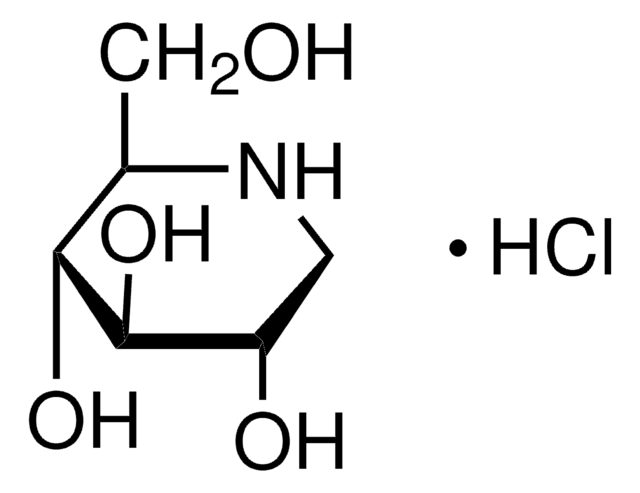
N-Butyldeoxynojirimycin
film (dried in situ), ≥98% (TLC)
Synonyme(s) :
Miglustat, NB-DNJ
About This Item
Produits recommandés

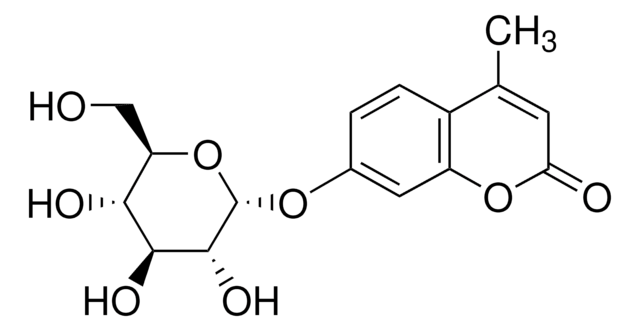
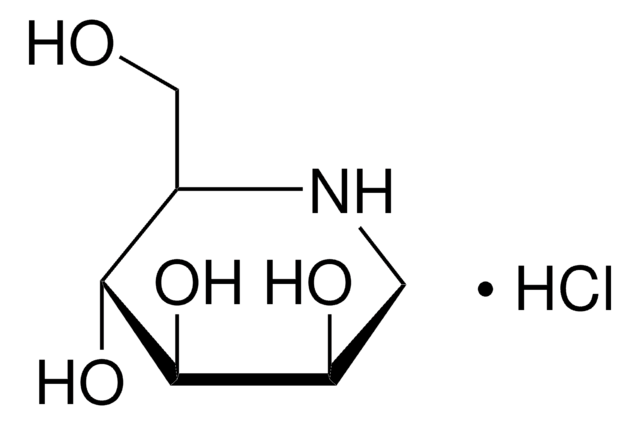

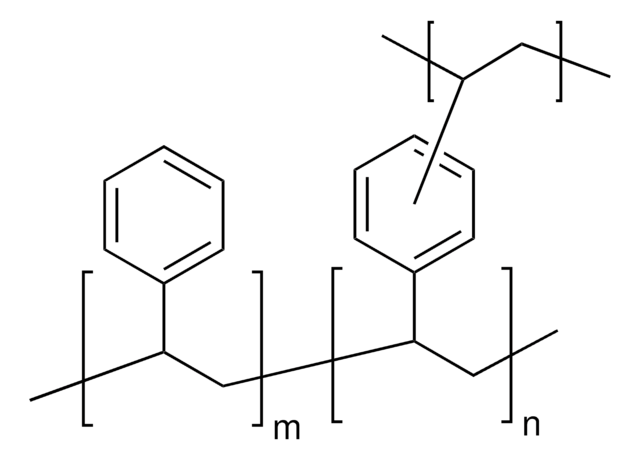
Nom du produit
N-Butyldeoxynojirimycin, film (dried in situ)
Essai
≥98% (TLC)
Niveau de qualité
Forme
film (dried in situ)
Solubilité
water: 9.80-10.20 mg/mL, clear, colorless
Température de stockage
2-8°C
Chaîne SMILES
CCCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO
InChI
1S/C10H21NO4/c1-2-3-4-11-5-8(13)10(15)9(14)7(11)6-12/h7-10,12-15H,2-6H2,1H3/t7-,8+,9-,10-/m1/s1
Clé InChI
UQRORFVVSGFNRO-UTINFBMNSA-N
Informations sur le gène
human ... UGCG(7357)
Catégories apparentées
Description générale
Application
- in the inhibition of glycolipid synthesis in neuroblastoma cells
- in the inhibition the ceramide-specific glycosyltransferase in hepatocytes
- in the inhibition of β-glucosidase (GBA2) using fluorescence- activity assay in human embryonic kidney (HEK293) cells.
Actions biochimiques/physiologiques
Code de la classe de stockage
11 - Combustible Solids
Classe de danger pour l'eau (WGK)
WGK 3
Point d'éclair (°F)
Not applicable
Point d'éclair (°C)
Not applicable
Équipement de protection individuelle
Eyeshields, Gloves, type N95 (US)
Faites votre choix parmi les versions les plus récentes :
Déjà en possession de ce produit ?
Retrouvez la documentation relative aux produits que vous avez récemment achetés dans la Bibliothèque de documents.
Les clients ont également consulté
Notre équipe de scientifiques dispose d'une expérience dans tous les secteurs de la recherche, notamment en sciences de la vie, science des matériaux, synthèse chimique, chromatographie, analyse et dans de nombreux autres domaines..
Contacter notre Service technique