
MBD6000
5R-Plex kit
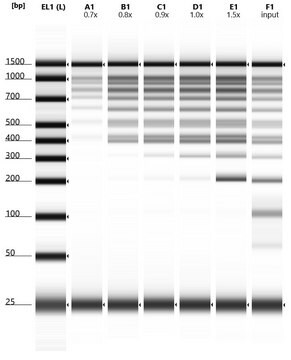
Ultra-sensitive 16S NGS assay for degraded and low biomass DNA
Sign Into View Organizational & Contract Pricing
All Photos(3)
About This Item
UNSPSC Code:
41106302
NACRES:
NA.84
Recommended Products








Quality Level
usage
Preparing 96 samples for sequencing
technique(s)
DNA amplification: suitable
DNA sequencing: suitable
shipped in
dry ice
storage temp.
-10 to -25°C
General description
The 16S ribosomal RNA gene (16S rRNA) is a common bacterial marker, used as a target for microbial community profiling in microbiome metagenomic studies. The gene is composed of nine variable regions (V1-V9) interspersed between conserved regions. The most common 16S rRNA NGS assays target one or two regions using a single set of primers (e.g., V3-V4). However, this approach usually results in a poor bacterial detection and classification rate when the DNA input is of low-quality or when applying it on extremely low biomass samples. The 5R-PLEX kit offers a complete solution to challenging biological samples where the standard 16S sequencing protocols fail to perform. Sample types include:
- Formalin-Fixed, Paraffin-Embedded (FFPE) tissue
- Cancerous tumor tissue
- Degraded or damaged DNA
- Low biomass samples
- Fossil-derived and ancient DNA
Features and Benefits
- The 5R-PLEX Kit provides high profile sequencing from degraded, damaged or low biomass samples
- Multiplexed NGS assay targets 5 variable regions of the 16S rRNA gene in a single primer pool
- Includes positive control for trouble shooting experiments.
- The kit can detect as low as 1pg of highly degraded bacterial DNA with an average fragment size of 260bp.
- Kit includes low bioburden reagents
- Includes exclusive access to M-CAMP™ platform featuring an innovative algorithm designed to reconstruct a single coherent microbial profiling and perform comprehensive analysis.
Components
- 5R-PLEX PCR1 Primer mix- 25 μL
- 5R-PLEX PCR2 Forward Primer mix- 25 μL
- 5R-PLEX index plate 96-plate
- Water, microbial DNA-free 10X- 1.5 mL
- HF DNA Polymerase- 0.1 mL
- 5X HF buffer 2 mL
- dNTP′s- 0.2 mL
- Elution Buffer (EB), microbial DNA-free- 8 mL
- 5R-PLEX Positive Control (10 ng/μL)- 30 μL
Storage and Stability
The shelf life of all reagents provided in the kit is 12 months when stored properly. Store all components at -20 °C.
Other Notes
Kit component details can be found in the User guide.
Legal Information
M-Camp is a trademark of Merck KGaA, Darmstadt, Germany
hcodes
pcodes
Hazard Classifications
Aquatic Chronic 3
Storage Class
10 - Combustible liquids
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Nicole M Davis et al.
Microbiome, 6(1), 226-226 (2018-12-19)
The accuracy of microbial community surveys based on marker-gene and metagenomic sequencing (MGS) suffers from the presence of contaminants-DNA sequences not truly present in the sample. Contaminants come from various sources, including reagents. Appropriate laboratory practices can reduce contamination, but
Jacob T Nearing et al.
Microbiome, 9(1), 113-113 (2021-05-20)
Advances in DNA sequencing technology have vastly improved the ability of researchers to explore the microbial inhabitants of the human body. Unfortunately, while these studies have uncovered the importance of these microbial communities to our health, they often do not
Deborah Nejman et al.
Science (New York, N.Y.), 368(6494), 973-980 (2020-05-30)
Bacteria were first detected in human tumors more than 100 years ago, but the characterization of the tumor microbiome has remained challenging because of its low biomass. We undertook a comprehensive analysis of the tumor microbiome, studying 1526 tumors and
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service