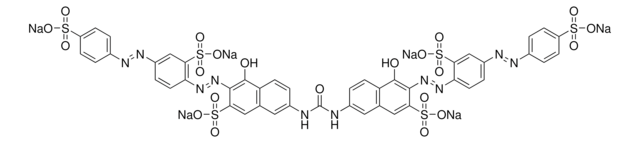
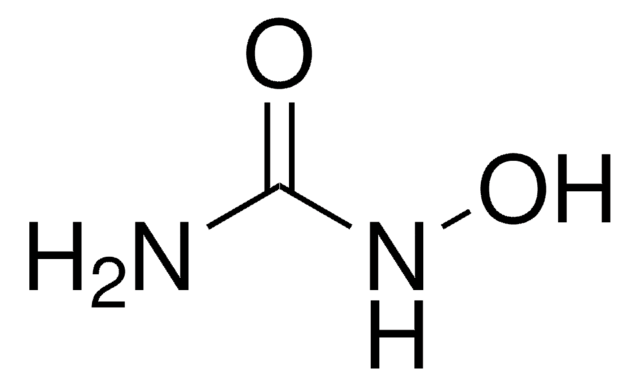
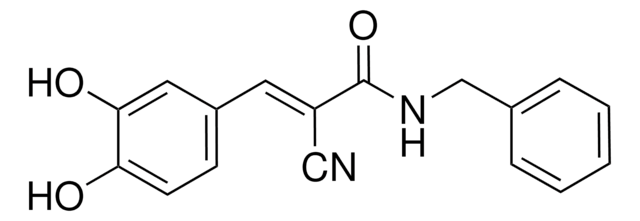
General description
THZ1 is a highly potent, selective and irreversible inhibitor of Cdk7 (IC50 = 3.2 nM). Acts by allosterically and covalently modifying Cys312 located outside the canonical kinase domain. Shown to block the phosphorylation of Ser5 and Ser7 in RNAPII CTD, the intracellular substrate of Cdk7 (<250 nM, 4 h in Jurkat cells). Does not affect the activity of C312S mutated Cdk7. Exhibits comparatively week activity towards Cdk12 (IC50 = 250 nM), Cdk1, and Cdk2. Suppresses the proliferation and cell viability of T-ALL and CLL cell lines and induces apoptosis with significant reduction in the levels of MCL-1 and XIAP. Also induces a significant reduction in the transcript and protein levels of RUNX1, TAL, and GATA3 (~ 50 nM). Suppresses the growth of KOPTK1 cells xenografts in murine model (10 mg/kg, b.i.d).
Biochem/physiol Actions
THZ1, an acrylamide-bearing phenylaminopyrimidine that, despite its ATP-competitive affinities toward multiple kinases (IC50 ≤1 µM against ATP probe binding to 22 kinases in a KiNativ panel screening), only effectively acts as an irreversible inhibitor against Cdk7 & Cdk12, and likely also Ckd13 & Cdk19, due to conformational positioning of a C-terminal Cys outside the kinase domain (C312 of human & murine Cdk7; C1539, C1517, C349 of human Cdk12, Cdk13, Cdk19, respectively) adjacent to the drug′s acrylamide moiety in the ATP cleft, allowing covalent bond formation. Due to the irreversible mode of action, 3 to 5 h incubation is shown to most effectively inhibit intracellular Ckd7 & Cdk12 (Effective conc. 100 to 500 nM in 293A, HeLa S3, Jurkat, and Loucy cultures). Likewise, drug washout following THZ1 incubation is demonstrated not to affect its potency against cellular Cdk7 substrates phosphorylation (RNAPII S2/5/7, Cdk1 T161, Cdk2 T160, Cdk9 T186) or cell proliferation. A strong enrichment of factors involved in Cdk7 substrate RNAPII/RPB1-driven transcriptional regulation is found among THZ1-sensitive cell lines with T cell acute lymphoblastic leukemia (T-ALL; IC50 in nM = 0.49/KOPTK1, 0.54/Loucy, 0.61/DND-41, 50 nM/Jurkat by cellular ATP measurement post 72 incubation) that depend on RUNX1 transcription for proliferation being particularly sensitive to THZ1-induced apoptotic death. Intravenous injection is efficacious in suppression KOPTK1 proliferation in mice (by 91%;10 mg/3.3 mL/Kg/12 h, BID, for 29 d one week following cancer cells injection) in vivo without affecting animal body weight.
Features and Benefits
- Cell-permeable
- Non-toxic
- ATP-site directed
- Irreversible inhibitor
Warning
Toxicity: Standard Handling (A)
Reconstitution
Following reconstitution, aliquot and freeze (-20°C). Stock solutions are stable for up to 3 months at -20°C.
Use only fresh DMSO for reconstitution.
Other Notes
Please note that the molecular weight for this compound is batch-specific due to variable water content.
Legal Information
CALBIOCHEM is a registered trademark of Merck KGaA, Darmstadt, Germany