
推荐产品
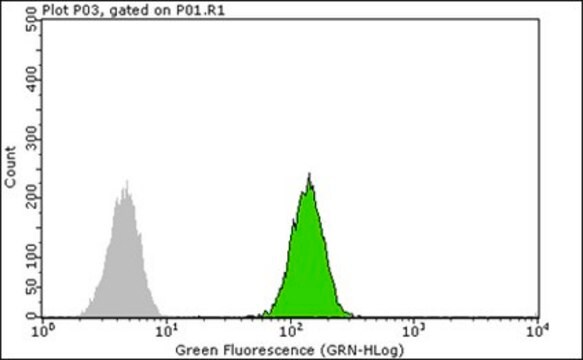
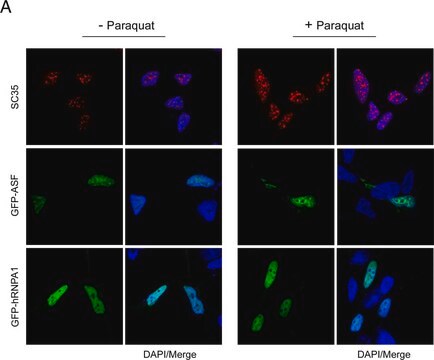




生物源
mouse
品質等級
共軛
unconjugated
抗體表格
purified from hybridoma cell culture
抗體產品種類
primary antibodies
無性繁殖
2B1, monoclonal
形狀
buffered aqueous solution
物種活性
Xenopus, human, mouse
濃度
~2 mg/mL
技術
microarray: suitable
western blot: 2-4 μg/mL using A431 cell extract
同型
IgG1
運輸包裝
dry ice
儲存溫度
−20°C
目標翻譯後修改
unmodified
基因資訊
human ... SMN1(6606) , SMN2(6607)
mouse ... Smn1(20595)
一般說明
Mouse monoclonal clone 2B1 anti-SMN antibody recognizes human, mouse, and Xenopus survival of motor neurons proteins.
特異性
The antibody recognizes human, mouse, and Xenopus SMN.
免疫原
recombinant human SMN
應用
Applications in which this antibody has been used successfully, and the associated peer-reviewed papers, are given below.
Enzyme-linked immunosorbent assay (1 paper)
Enzyme-linked immunosorbent assay (1 paper)
Mouse monoclonal clone 2B1 anti-SMN antibody is an important tool for studying the role of the survival of motor neurons protein in nuclear processes and spinal muscular atrophy (SMA). It may be used in immunoblotting (~35 kDa), immunoprecipitation, and immunocytochemistry.
生化/生理作用
Survival of Motor Neurons (SMN) complex is important in various biological processes, such as assembly and restructuring of spliceosomal small nuclear ribonucleoproteins (snRNPs), pre mRNA splicing and transcription. Spinal muscular atrophy (SMA) is caused by reduced expression or mutations in the survival of motor neurons (SMN) protein. Deletion or mutation in the telomeric copy (SMN1) causes the SMA phenotype. The severity of SMA is in direct correlation with the expression level of the SMN protein, either from the SMN1 gene or a different spliced form of SMN from the SMN2 gene. The SMN complex interacts with various protein substrates such as Sm and Lsm proteins of the spliceosomal snRNPs, fibrillarin, GAR1, RNA helicase A, the human hnRNP proteins (hnRNPQ, U and R), coilin and p53.
外觀
Solution in 0.01 M phosphate buffered saline, pH 7.4, containing 15mM sodium azide.
儲存和穩定性
For continuous use, store at 2-8 °C for up to one month. For extended storage, freeze in working aliquots. Repeated freezing and thawing, or storage in "frostfree" freezers, is not recommended. If slight turbidity occurs upon prolonged storage, clarify the solution by centrifugation before use. Working dilution samples should be discarded if not used within 12 hours.
免責聲明
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
未找到合适的产品?
试试我们的产品选型工具.
儲存類別代碼
12 - Non Combustible Liquids
水污染物質分類(WGK)
WGK 2
閃點(°F)
Not applicable
閃點(°C)
Not applicable
Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?
Burghes AHM and Beattie CE
Nature Reviews. Neuroscience, 10(8), 597-597 (2009)
Dione T Kobayashi et al.
PloS one, 6(8), e24269-e24269 (2011-09-10)
Genetic defects leading to the reduction of the survival motor neuron protein (SMN) are a causal factor for Spinal Muscular Atrophy (SMA). While there are a number of therapies under evaluation as potential treatments for SMA, there is a critical
Q Liu et al.
The EMBO journal, 15(14), 3555-3565 (1996-07-15)
Spinal muscular atrophy (SMA) is a common, often fatal, autosomal recessive disease leading to progressive muscle wasting and paralysis as a result of degeneration of anterior horn cells of the spinal cord. A gene termed survival of motor neurons (SMN)
Thomas O Crawford et al.
PloS one, 7(4), e33572-e33572 (2012-05-05)
The universal presence of a gene (SMN2) nearly identical to the mutated SMN1 gene responsible for Spinal Muscular Atrophy (SMA) has proved an enticing incentive to therapeutics development. Early disappointments from putative SMN-enhancing agent clinical trials have increased interest in
Sergey Paushkin et al.
Current opinion in cell biology, 14(3), 305-312 (2002-06-18)
Spinal muscular atrophy is a common, often lethal, neurodegenerative disease that results from low levels of, or loss-of-function mutations in, the SMN (survival of motor neurons) protein. SMN oligomerizes and forms a stable complex with five additional proteins: Gemins 2-6.
我们的科学家团队拥有各种研究领域经验,包括生命科学、材料科学、化学合成、色谱、分析及许多其他领域.
联系技术服务部门