
05-583
Anti-CFTR-Antikörper, Klon M3A7
clone M3A7, Upstate®, from mouse
Synonym(e):
ATP-binding cassette sub-family C, member 7, ATP-binding cassette transporter sub-family C member 7, cAMP-dependent chloride channel, cystic fibrosis transmembrane conductance regulator, cystic fibrosis transmembrane conductance regulator (ATP-binding ca
About This Item
Empfohlene Produkte





Biologische Quelle
mouse
Qualitätsniveau
Antikörperform
purified immunoglobulin
Antikörper-Produkttyp
primary antibodies
Klon
M3A7, monoclonal
Speziesreaktivität
human
Hersteller/Markenname
Upstate®
Methode(n)
immunohistochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
Isotyp
IgG1
NCBI-Hinterlegungsnummer
UniProt-Hinterlegungsnummer
Versandbedingung
dry ice
Posttranslationale Modifikation Target
unmodified
Angaben zum Gen
human ... CFTR(1080)
Allgemeine Beschreibung
Spezifität
Immunogen
Anwendung
Neurowissenschaft
Ionenkanäle & -transporter
Dieser Antikörper immunpräzipitiert CFTR. (Kartner, N., 1998.)
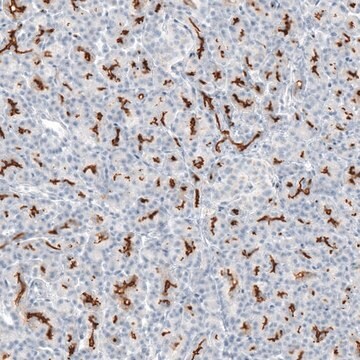
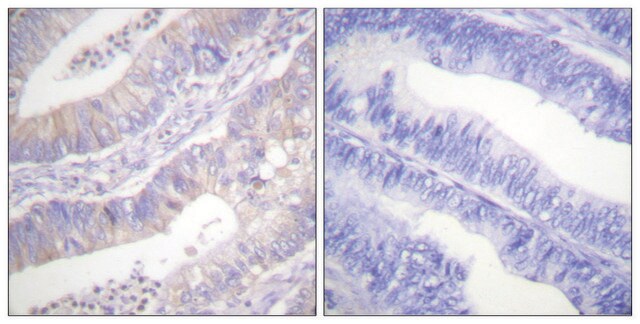
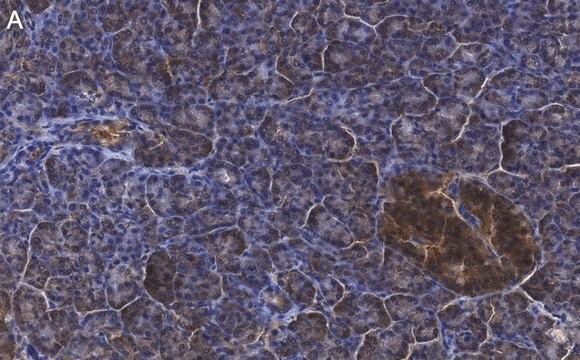
Immunohistochemie:
Dieser Antikörper immunfärbt CFTR in Gewebeschnitten von humanem Pankreas. (Kartner, N., 1998.)
Qualität
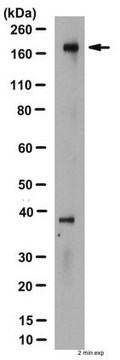
Western-Blot-Analyse:
0,5–2 µg/ml dieser Charge wiesen CFTR in 20–50 µg RIPA-Zelllysaten von humanem T84-Darmkarzinom-Epithel nach. 0,5–2 µg/ml einer früheren Charge wiesen CFTR in mit CFTR transfizierten BHK nach (Haardt, M., 1999)
Hinweis: Sieden Sie das Lysat nicht. Stattdessen bei 37 °C 30 Minuten inkubieren. CFTR kann als diffundiertes Protein auf SDS-PAGE laufen.
Zielbeschreibung
Physikalische Form
Lagerung und Haltbarkeit
Handhabungsempfehlungen:
Das Fläschchen nach Empfang und vor dem Abnehmen der Kappe zentrifugieren und die Lösung vorsichtig mischen. In Mikrozentrifugenröhrchen aliquotieren und bei -20 °C aufbewahren. Wiederholte Einfrier-/Auftauzyklen sind zu vermeiden, da sie das IgG beschädigen und die Produktleistung beeinträchtigen können. Hinweis: Ein Abfallen der Gefrierschranktemperatur unter -20 °C kann dazu führen, dass glycerinhaltige Lösungen während der Lagerung einfrieren.
Hinweis zur Analyse
T84-Zelllysat.
Sonstige Hinweise
Rechtliche Hinweise
Haftungsausschluss
Sie haben nicht das passende Produkt gefunden?
Probieren Sie unser Produkt-Auswahlhilfe. aus.
Empfehlung
Lagerklassenschlüssel
10 - Combustible liquids
WGK
WGK 1
Analysenzertifikate (COA)
Suchen Sie nach Analysenzertifikate (COA), indem Sie die Lot-/Chargennummer des Produkts eingeben. Lot- und Chargennummern sind auf dem Produktetikett hinter den Wörtern ‘Lot’ oder ‘Batch’ (Lot oder Charge) zu finden.
Besitzen Sie dieses Produkt bereits?
In der Dokumentenbibliothek finden Sie die Dokumentation zu den Produkten, die Sie kürzlich erworben haben.
Unser Team von Wissenschaftlern verfügt über Erfahrung in allen Forschungsbereichen einschließlich Life Science, Materialwissenschaften, chemischer Synthese, Chromatographie, Analytik und vielen mehr..
Setzen Sie sich mit dem technischen Dienst in Verbindung.