
05-583
Anticorpo anti-CFTR, clone M3A7
clone M3A7, Upstate®, from mouse
Sinonimo/i:
ATP-binding cassette sub-family C, member 7, ATP-binding cassette transporter sub-family C member 7, cAMP-dependent chloride channel, cystic fibrosis transmembrane conductance regulator, cystic fibrosis transmembrane conductance regulator (ATP-binding ca
About This Item
Prodotti consigliati





Origine biologica
mouse
Livello qualitativo
Forma dell’anticorpo
purified immunoglobulin
Tipo di anticorpo
primary antibodies
Clone
M3A7, monoclonal
Reattività contro le specie
human
Produttore/marchio commerciale
Upstate®
tecniche
immunohistochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
Isotipo
IgG1
N° accesso NCBI
N° accesso UniProt
Condizioni di spedizione
dry ice
modifica post-traduzionali bersaglio
unmodified
Informazioni sul gene
human ... CFTR(1080)
Descrizione generale
Specificità
Immunogeno
Applicazioni
Neuroscienze
questo anticorpo è stato descritto in saggi di immunoprecipitazione del CFTR. (Kartner, N., 1998.)
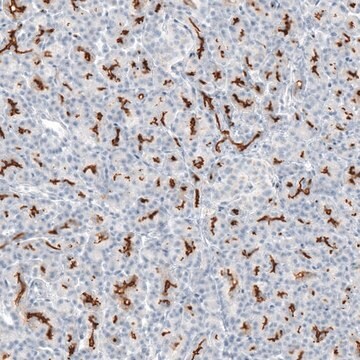
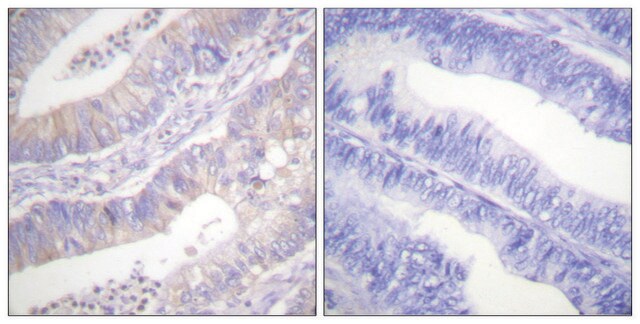
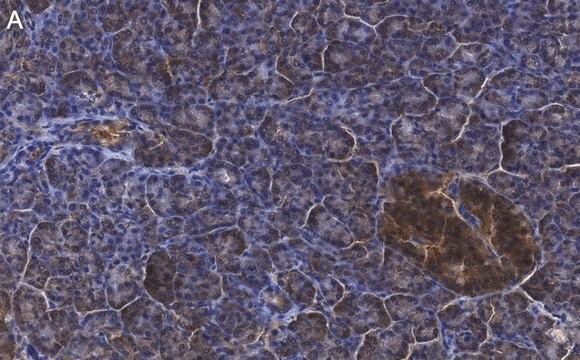
Immunoistochimica:
questo anticorpo è stato descritto il lavori di immunocolorazione del CFTR in sezioni di tessuto pancreatico umano. (Kartner, N., 1998).
Canali & pompe ioniche
Qualità
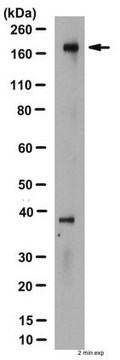
Western blotting:
una concentrazione compresa tra 0,5 e 2 µg/mL di un lotto rappresentativo ha permesso di individuare il CFTR in 20-50 µg di lisati in tampone per RIPA di cellule epiteliali di carcinoma di colon umano T84. Una concentrazione compresa tra 0,5 e 2 µg/mL di un lotto rappresentativo ha permesso di individuare il CFTR in cellule BHK trasfettate con CFTR (Haardt, M., 1999).
N.B.: Non bollire i lisati. Incubarli a 37 °C per 30 minuti. In SDS-PAGE il CFTR può migrare come una banda diffusa.
Descrizione del bersaglio
Stato fisico
Stoccaggio e stabilità
Indicazioni per l'uso:
una volta ricevuto il prodotto, prima di rimuovere il tappo, centrifugare la provetta e mescolare delicatamente la soluzione. Suddividere in aliquote in provette per microcentrifuga e conservare a -20 °C. Evitare ripetuti cicli di congelamento e scongelamento che potrebbero danneggiare le IgG e compromettere le prestazioni del prodotto. N.B: oscillazioni di temperatura del congelatore al di sotto dei -20°C possono causare il congelamento delle soluzioni contenenti glicerolo.
Risultati analitici
Lisato di cellule T84
Altre note
Note legali
Esclusione di responsabilità
Non trovi il prodotto giusto?
Prova il nostro Motore di ricerca dei prodotti.
Raccomandato
Codice della classe di stoccaggio
10 - Combustible liquids
Classe di pericolosità dell'acqua (WGK)
WGK 1
Certificati d'analisi (COA)
Cerca il Certificati d'analisi (COA) digitando il numero di lotto/batch corrispondente. I numeri di lotto o di batch sono stampati sull'etichetta dei prodotti dopo la parola ‘Lotto’ o ‘Batch’.
Possiedi già questo prodotto?
I documenti relativi ai prodotti acquistati recentemente sono disponibili nell’Archivio dei documenti.
Il team dei nostri ricercatori vanta grande esperienza in tutte le aree della ricerca quali Life Science, scienza dei materiali, sintesi chimica, cromatografia, discipline analitiche, ecc..
Contatta l'Assistenza Tecnica.