
Wichtige Dokumente
43659
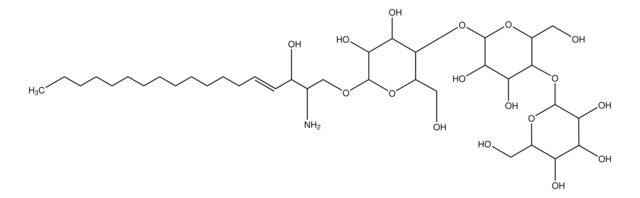
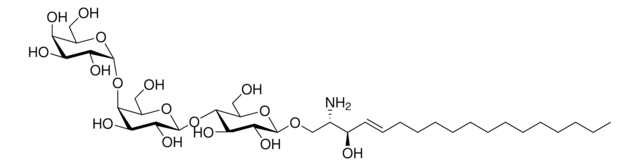
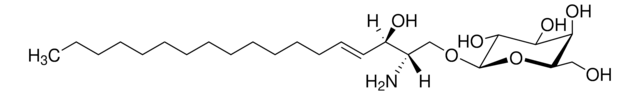
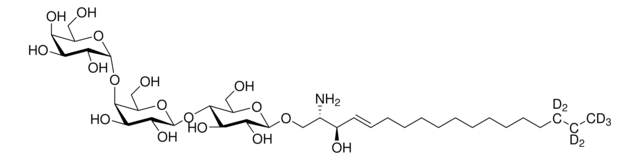
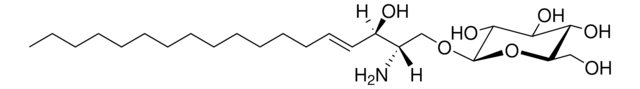
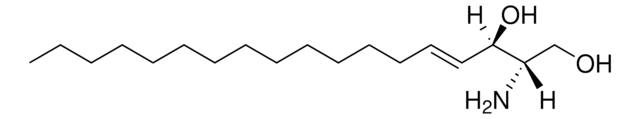
Glucosylsphingosin
≥98.0% (TLC)
Synonym(e):
(2S,3R,4E)-2-Amino-3-hydroxy-4-octadecen-1-yl β-D-glucopyranosid, 1-β-D-Glucosylsphingosin, Glucosyl-C18-sphingosin
About This Item
Empfohlene Produkte


Qualitätsniveau
Assay
≥98.0% (TLC)
Form
powder
Lipid-Typ
sphingolipids
Lagertemp.
−20°C
SMILES String
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChIKey
HHJTWTPUPVQKNA-JLRUQHRASA-N
Biochem./physiol. Wirkung
Lagerklassenschlüssel
11 - Combustible Solids
WGK
WGK 3
Flammpunkt (°F)
Not applicable
Flammpunkt (°C)
Not applicable
Hier finden Sie alle aktuellen Versionen:
Besitzen Sie dieses Produkt bereits?
In der Dokumentenbibliothek finden Sie die Dokumentation zu den Produkten, die Sie kürzlich erworben haben.
Kunden haben sich ebenfalls angesehen
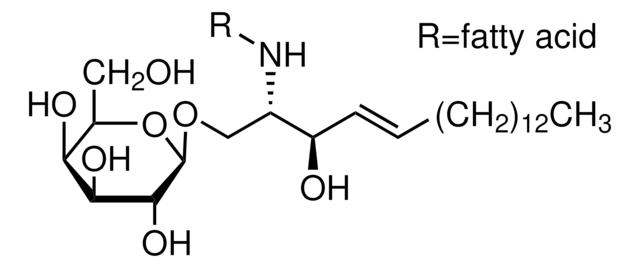

Unser Team von Wissenschaftlern verfügt über Erfahrung in allen Forschungsbereichen einschließlich Life Science, Materialwissenschaften, chemischer Synthese, Chromatographie, Analytik und vielen mehr..
Setzen Sie sich mit dem technischen Dienst in Verbindung.