
推荐产品


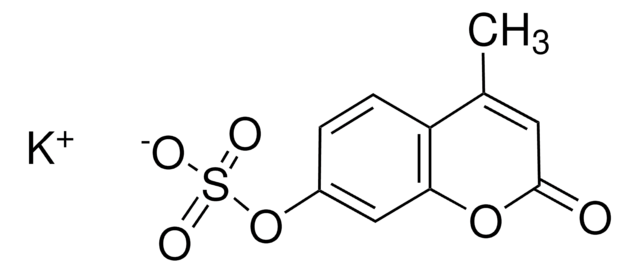
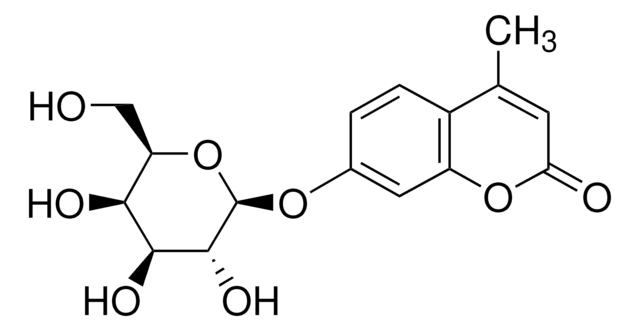
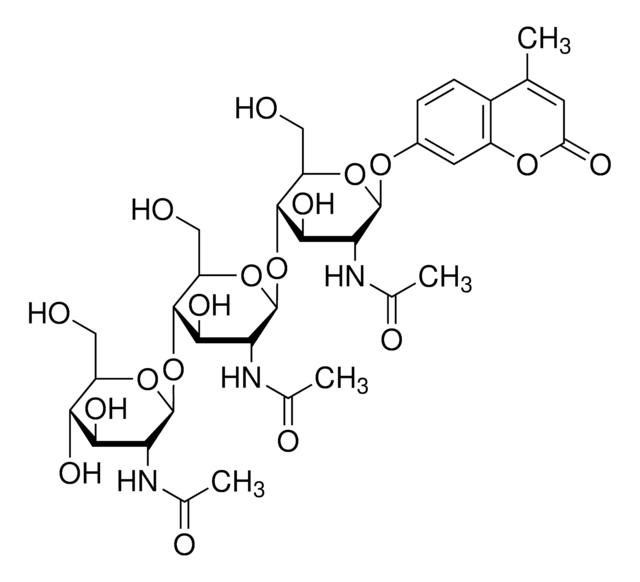
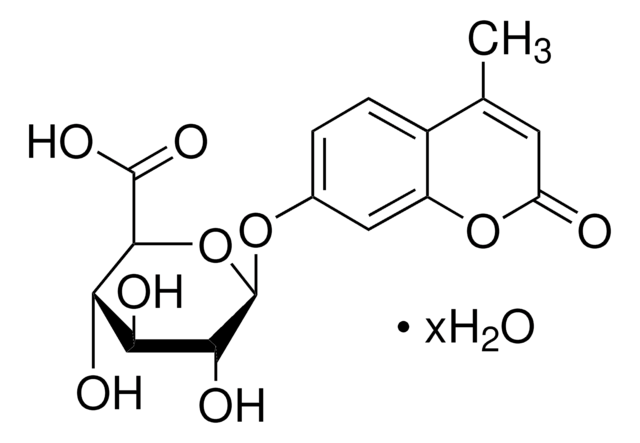
重組細胞
expressed in mouse NSO cells
品質等級
形狀
solution
比活性
≥7,500 units/μg protein
分子量
83 kDa
雜質
≤1.0 EU/μg Endotoxin
運輸包裝
wet ice
儲存溫度
−20°C
一般說明
α-L-艾杜糖苷酸酶 (IDUA) 定位于人染色体4p16.3。成熟的IDUA蛋白被糖基化,并包含三糖磷酸异构酶(TIM)桶状结构域、β夹心式螺旋-环-螺旋区域和免疫球蛋白样结构域。α-L-艾杜糖苷酸酶被归类为糖苷水解酶(GH)家族39。
應用
α-L-艾杜糖醛酸酶可用于新生儿 a-L-艾杜糖醛酸酶缺乏症的白细胞测定。
生化/生理作用
α-L-艾杜糖苷酸酶突变与粘多糖贮积病I型(MPS I)有关。该酶的缺陷导致皮肤素和硫酸乙酰肝素的积累。MPS I的病理生理学伴随着颅骨变形、智力低下和疝气。
在溶酶体降解过程中,α-L-艾杜糖苷酸酶(Iduronidase )起着至关重要的作用。它水解糖胺聚糖(GAG)(包括硫酸皮肤素和硫酸乙酰肝素) 中非还原末端的α L-艾杜糖醛酸残基。
催化硫酸皮肤素中未硫酸化的 L -艾杜糖苷键的水解
物理性質
在 SDS-PAGE 还原条件下,表达为 C-末端组氨酸标记蛋白(残基 1-653),其含钙核分子量为 71 kDa,在约 83 kDa 处迁移。
單位定義
一个单元将在-25°C pH 3.5 下每分钟从 4-甲基伞形酮-α-L-艾杜糖苷酸生成 1 皮摩尔的 4-甲基伞形酮。
外觀
40 mM 醋酸钠、400 mM NaCl 和 20% (v/v) 甘油溶液 (pH 5.0)
儲存類別代碼
10 - Combustible liquids
水污染物質分類(WGK)
WGK 1
閃點(°F)
Not applicable
閃點(°C)
Not applicable
其他客户在看


Kristin D'Aco et al.
European journal of pediatrics, 171(6), 911-919 (2012-01-12)
Our objective was to assess how the diagnosis and treatment of mucopolysaccharidosis I (MPS I) have changed over time. We used data from 891 patients in the MPS I Registry, an international observational database, to analyze ages at symptom onset
Akemi Tanaka et al.
Molecular genetics and metabolism, 107(3), 513-520 (2012-10-02)
Hematopoietic stem cell transplantation (HSCT) has not been indicated for patients with mucopolysaccharidosis II (MPS II, Hunter syndrome), while it is indicated for mucopolysaccharidosis I (MPS I) patients <2 years of age and an intelligence quotient (IQ) of ≥ 70.
Ruben J Boado et al.
Bioconjugate chemistry, 24(1), 97-104 (2012-12-20)
The chronic administration of recombinant fusion proteins in preclinical animal models may generate an immune response and the formation of antidrug antibodies (ADA). Such ADAs could alter the plasma pharmacokinetics of the fusion protein, and mask any underlying toxicity of
Minke H de Ru et al.
Orphanet journal of rare diseases, 7, 22-22 (2012-04-25)
Mucopolysaccharidosis type I (MPS I) is traditionally divided into three phenotypes: the severe Hurler (MPS I-H) phenotype, the intermediate Hurler-Scheie (MPS I-H/S) phenotype and the attenuated Scheie (MPS I-S) phenotype. However, there are no clear criteria for delineating the different
Xu He et al.
Nature communications, 3, 1062-1062 (2012-09-20)
Lysosomal storage diseases are a class of over 70 rare genetic diseases that are amenable to enzyme replacement therapy. Towards developing a plant-based enzyme replacement therapeutic for the lysosomal storage disease mucopolysaccharidosis I, here we expressed α-L-iduronidase in the endosperm
我们的科学家团队拥有各种研究领域经验,包括生命科学、材料科学、化学合成、色谱、分析及许多其他领域.
联系技术服务部门