
推荐产品


品質等級
化驗
≥98.0% (TLC)
形狀
powder
脂質類型
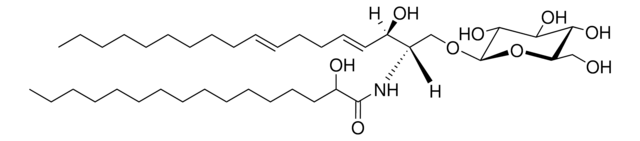
sphingolipids
儲存溫度
−20°C
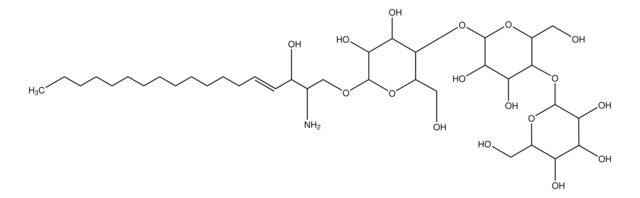
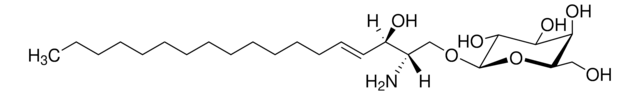
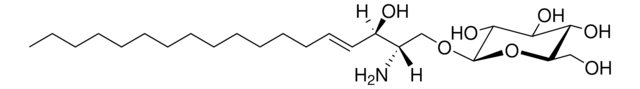
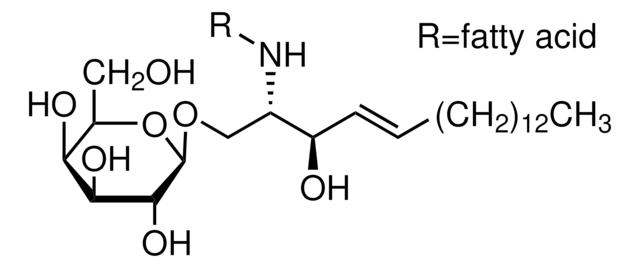
SMILES 字串
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChI 密鑰
HHJTWTPUPVQKNA-JLRUQHRASA-N
生化/生理作用
Glucosylsphingosine is a cytotoxic compound. Accumulation of glucosylsphingosine in brain and other tissues occurs in patients with Gaucher disease, which is an inherited deficiency of lysosomal glucocerebrosidase, which converts glucosylsphingosine to glucose and sphingosine.
儲存類別代碼
11 - Combustible Solids
水污染物質分類(WGK)
WGK 3
閃點(°F)
Not applicable
閃點(°C)
Not applicable
其他客户在看
E M Kaye et al.
Annals of neurology, 20(2), 223-230 (1986-08-01)
Glucocerebroside levels were measured in the brains of patients with neuronopathic forms (types 2 and 3) of Gaucher disease and compared to those obtained from control brain. Nine separate brain regions (frontal, temporal, occipital, and cerebellar cortices; thalamus; corpus striatum;
A convenient approach to facilitate monitoring Gaucher disease progression and therapeutic response.
Wujuan Zhang et al.
The Analyst, 142(18), 3380-3387 (2017-08-16)
Gaucher disease (GD) is caused by mutations on the GBA1 gene leading to deficiency in acid β-glucosidase (GCase) and subsequent accumulation of its substrates, glucosylceramide (GlcC) and glucosylsphingosine (GlcS). GlcS in plasma has been proposed as a highly sensitive and
Chengfang Tang et al.
Clinical biochemistry, 87, 79-84 (2020-11-15)
Gaucher disease (GD) is caused by a deficiency of β-glucosidase (GCase), leading to accumulation of glucosylceramide (GlcC) and glucosylsphingosine (Lyso-Gb1). Lyso-Gb1 is a reliable biomarker for GD. This study aims to develop a simple, effective and accurate method for the
Ellen Sidransky
Molecular genetics and metabolism, 83(1-2), 6-15 (2004-10-07)
Gaucher disease, the recessively inherited deficiency of the enzyme glucocerebrosidase and the most common sphingolipidosis, has both non-neurological and neuronopathic forms and a continuum of diverse clinical manifestations. Studies of genotype-phenotype correlations reveal significant genotypic heterogeneity among clinically similar patients
E Beutler
Blood reviews, 2(1), 59-70 (1988-03-01)
Gaucher disease is a glycolipid storage disorder characterized by accumulation of glucocerebroside in the liver, spleen, and bones, and caused by a deficiency of glucocerebrosidase. Glucocerebrosidase cDNA has been cloned and sequenced, and much has been learned about the synthesis
我们的科学家团队拥有各种研究领域经验,包括生命科学、材料科学、化学合成、色谱、分析及许多其他领域.
联系技术服务部门