
MAK320
D-2-Hydroxyglutarate (D2HG) Assay Kit
sufficient for 200 fluorometric reactions
Synonym(s):
D2HG Test Kit
Sign Into View Organizational & Contract Pricing
All Photos(1)
About This Item
UNSPSC Code:
12161503
NACRES:
NA.84
Recommended Products


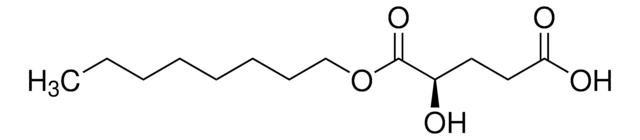
usage
sufficient for 200 reactions (Fluorometric)
detection method
fluorometric
relevant disease(s)
cancer; neurological disorders
shipped in
wet ice
storage temp.
−20°C
General description
The level of D-2-Hydroxyglutarate (D2HG) is low in normal cells and tissues, but is significantly elevated in metabolic diseases, such as the rare autosomal disorder D2HG aciduria. D2HG is mildly elevated in other metabolic disorders including multiple acyl-CoA dehydrogenase deficiency, dihydrolipoyl dehydrogenase deficiency, pyruvate decarboxylase deficiency and pyruvate carboxylase deficiency, various cancers, and in neoplasms with mutations in the isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) genes. Detection of elevated D2HG is an important biomarker for early diagnosis, prognosis, monitoring and the development of therapeutic strategies against these diseases.
Features and Benefits
- Highly sensitive and rapid enzymatic assay for the detection of D2HG levels in various biological fluids
- Supportive calculator (Click here to download a calculator excel file) Analyze results based on your experimental data!
- Quick instruction bench card - to assure your experimental success
- More experiments in one kit - contains sufficient reagents for 200 tests
- Detection time: only 30-60 minutes
Suitability
Suitable for the detection of D2HG levels in cells and tissues lysates, serum, urine, cultured cells and culture supernatants.
Principle
Developed in partnership with the German Cancer Research Center (DKFZ), the D2HG Assay Kit is a rapid and sensitive enzymatic assay for the detection of D2HG levels in various biological fluids: serum, urine, cell culture supernatants, and cell or tissue lysates. The assay, originally developed by Balss et al., 8 is based on the oxidation of D2HG to α-ketoglutarate (αKG) by the enzyme (D)-2-hydroxyglutarate dehydrogenase (HGDH) coupled to the reduction of NAD+ to NADH (see Figure 1). The amount of NADH formed is then quantitated by the diaphorase mediated reduction of resazurin to the fluorescent dye resorufin (λex = 540 nm/λem = 590 nm).
Legal Information
This product is sold under license from the German Cancer Research Center (DKFZ) and University Clinic of Heidelberg. Use of this product is covered by certain US and foreign patents, including US Pat. No. 9,487,815, EP Pat. No. 2 820 145 and foreign equivalents. Use of this product is for research purposes only.
Storage Class Code
11 - Combustible Solids
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Customers Also Viewed

R A Chalmers et al.
Journal of inherited metabolic disease, 3(1), 11-15 (1980-01-01)
A patient with protein-losing gastroenteropathy and egg allergy has been shown to have a previously unrecognized organic aciduria, D-2-hydroxyglutaric aciduria. The observations made are consistent with an inherited metabolic disorder in the catabolism of 5-aminolaevulinate possibly due to deficient activity
Roberta Leonardi et al.
The Journal of biological chemistry, 287(18), 14615-14620 (2012-03-24)
Isocitrate dehydrogenase (IDH) is a reversible enzyme that catalyzes the NADP(+)-dependent oxidative decarboxylation of isocitrate (ICT) to α-ketoglutarate (αKG) and the NADPH/CO(2)-dependent reductive carboxylation of αKG to ICT. Reductive carboxylation by IDH1 was potently inhibited by NADP(+) and, to a
Eduard A Struys et al.
American journal of human genetics, 76(2), 358-360 (2004-12-21)
d-2-hydroxyglutaric aciduria is a neurometabolic disorder with both a mild and a severe phenotype and with unknown etiology. Recently, a novel enzyme, d-2-hydroxyglutarate dehydrogenase, which converts d-2-hydroxyglutarate into 2-ketoglutarate, and its gene were identified. In the genes of two unrelated
R A Chalmers et al.
Clinica chimica acta; international journal of clinical chemistry, 77(2), 117-124 (1977-06-01)
Detailed studies, using gas chromatography and mass spectrometric methods, of the urinary organic acids excreted by a patient with proven pyruvate decarboxylase deficiency are reported. In addition to the greatly-increased levels of lactate and pyruvate, marked elevation in the levels
S I Goodman et al.
Pediatric research, 14(1), 12-17 (1980-01-01)
When amino acids were infused at a rate of 4 g/kg/day, an infant with hypoglycemia, metabolic acidemia and chronic regurgitation showed hypersarcosinemia and excreted abnormal amounts of sarcosine, isovalerylglycine, isobutyrylglycine, alpha-methylbutyrylglycine, and beta-hydroxyisovaleric, glutaric, alpha-hydroxyglutaric, methylsuccinic, and alpha-hydroxyisobutyric acids in
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service