
S2944
Anti-SMN antibody, Mouse monoclonal
clone 2B1, purified from hybridoma cell culture
Sinónimos:
Anti-Survival of Motor Neurons
About This Item
Productos recomendados




origen biológico
mouse
conjugado
unconjugated
forma del anticuerpo
purified from hybridoma cell culture
tipo de anticuerpo
primary antibodies
clon
2B1, monoclonal
formulario
buffered aqueous solution
reactividad de especies
Xenopus, human, mouse
concentración
~2 mg/mL
técnicas
microarray: suitable
western blot: 2-4 μg/mL using A431 cell extract
isotipo
IgG1
Condiciones de envío
dry ice
temp. de almacenamiento
−20°C
modificación del objetivo postraduccional
unmodified
Información sobre el gen
human ... SMN1(6606) , SMN2(6607)
mouse ... Smn1(20595)
Descripción general
Especificidad
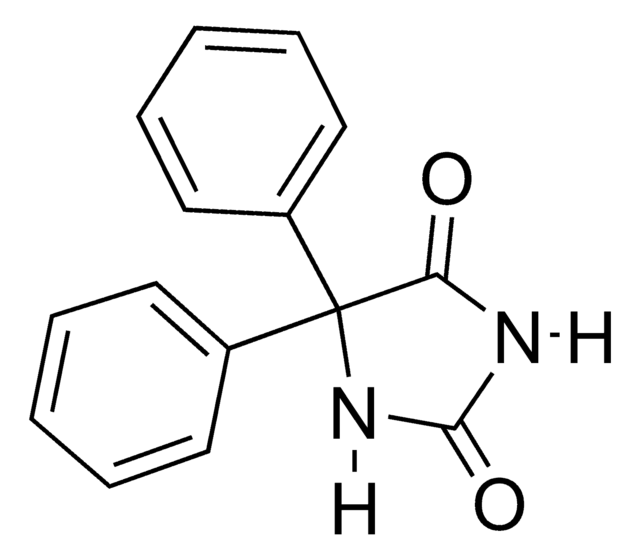
Inmunógeno
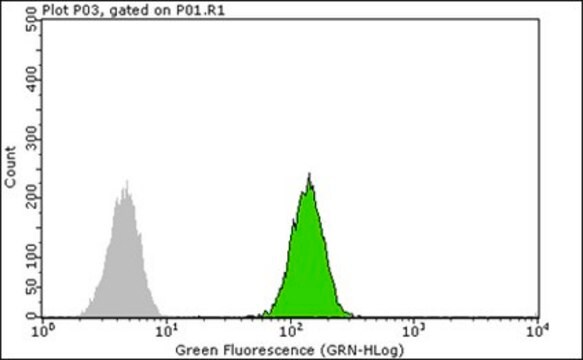
Aplicación
Enzyme-linked immunosorbent assay (1 paper)
Acciones bioquímicas o fisiológicas
Forma física
Almacenamiento y estabilidad
Cláusula de descargo de responsabilidad
¿No encuentra el producto adecuado?
Pruebe nuestro Herramienta de selección de productos.
Código de clase de almacenamiento
12 - Non Combustible Liquids
Clase de riesgo para el agua (WGK)
WGK 2
Punto de inflamabilidad (°F)
Not applicable
Punto de inflamabilidad (°C)
Not applicable
Certificados de análisis (COA)
Busque Certificados de análisis (COA) introduciendo el número de lote del producto. Los números de lote se encuentran en la etiqueta del producto después de las palabras «Lot» o «Batch»
¿Ya tiene este producto?
Encuentre la documentación para los productos que ha comprado recientemente en la Biblioteca de documentos.
Nuestro equipo de científicos tiene experiencia en todas las áreas de investigación: Ciencias de la vida, Ciencia de los materiales, Síntesis química, Cromatografía, Analítica y muchas otras.
Póngase en contacto con el Servicio técnico