
43659
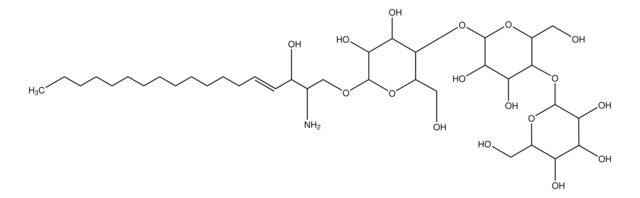
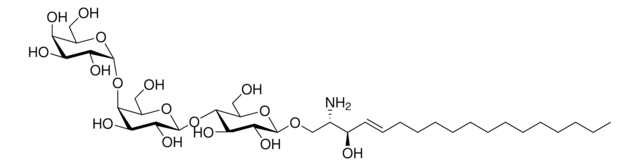
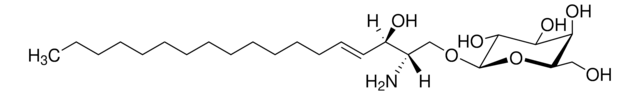
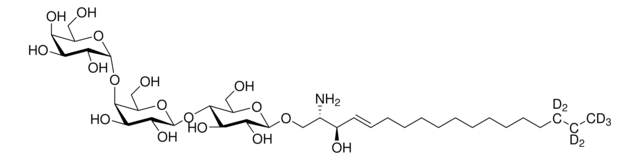
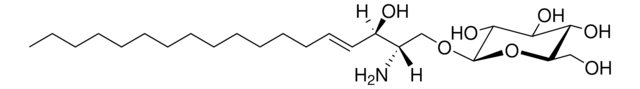
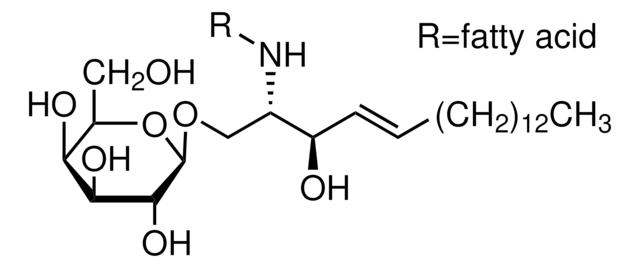
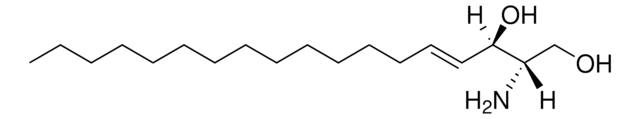
Glucosylsphingosine
≥98.0% (TLC)
Synonym(s):
(2S,3R,4E)-2-Amino-3-hydroxy-4-octadecen-1-yl β-D-glucopyranoside, 1-β-D-Glucosylsphingosine, Glucosyl-C18-sphingosine
About This Item
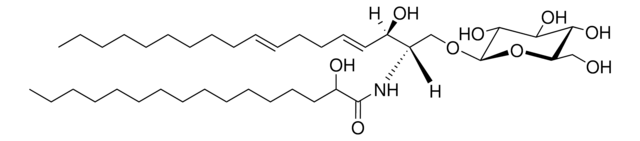
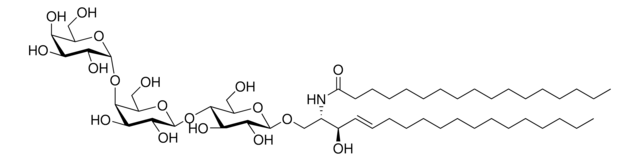
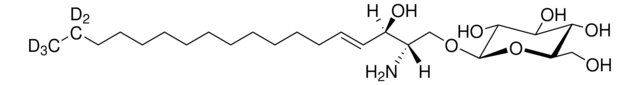
Recommended Products


Quality Level
Assay
≥98.0% (TLC)
form
powder
lipid type
sphingolipids
storage temp.
−20°C
SMILES string
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChI key
HHJTWTPUPVQKNA-JLRUQHRASA-N
Biochem/physiol Actions
Storage Class Code
11 - Combustible Solids
WGK
WGK 3
Flash Point(F)
Not applicable
Flash Point(C)
Not applicable
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Customers Also Viewed

Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service