
Wichtige Dokumente
UPS1
Universelles Proteomics-Standardset
Protein Mass Spectrometry Calibration Standard
Synonym(e):
Standardset
About This Item
Empfohlene Produkte






Form
ready-to-use solution
Qualitätsniveau
Qualität
Protein Mass Spectrometry Calibration Standard
Methode(n)
mass spectrometry (MS): suitable
Lagertemp.
−20°C
Allgemeine Beschreibung
Anwendung
- Bracketing von kritischen experimentellen Datensätzen, um die Robustheit von Analysemethoden zu bestätigen
- Vergleich von MS- oder anderen proteomischen Daten, die in verschiedenen Laboren mit einer Vielzahl von Analysestrategien und -geräten erzeugt werden
- Identifizierung der Beschränkungen von Systemen und Suchalgorithmen für proteomische Analysen
- Externe Referenz zur Unterstützung der Evaluierung von Daten, die aus schlecht definierten Proben gewonnen wurden
Leistungsmerkmale und Vorteile
- Überprüfen der Leistungskraft Ihrer Analysestrategie
- Fehlerbehebung und Optimierung bei Analyseprotokollen
- Bestätigen der Eignung eines Systems vor dem Analysieren kritischer Proben
- Normalisieren von Analyseergebnissen von Tag zu Tag und von Labor zu Labor
Kit-Komponenten auch einzeln erhältlich
- T6567Trypsin from porcine pancreas, Proteomics Grade, BioReagent, Dimethylated 20 μgSDB
Ähnliches Produkt



Signalwort
Danger
Gefahreneinstufungen
Acute Tox. 4 Oral - Eye Dam. 1 - Repr. 1B - Resp. Sens. 1 - Skin Irrit. 2 - STOT SE 3
Zielorgane
Respiratory system
Lagerklassenschlüssel
6.1C - Combustible acute toxic Cat.3 / toxic compounds or compounds which causing chronic effects
WGK
WGK 3
Hier finden Sie alle aktuellen Versionen:
Besitzen Sie dieses Produkt bereits?
In der Dokumentenbibliothek finden Sie die Dokumentation zu den Produkten, die Sie kürzlich erworben haben.
Kunden haben sich ebenfalls angesehen






Artikel
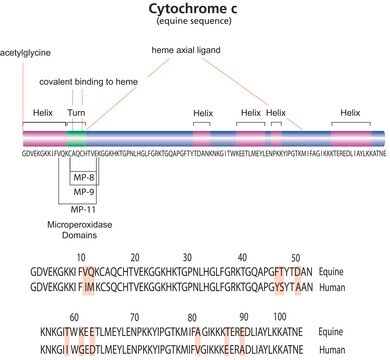
The era of high-throughput proteomics has recently blossomed due in large part to advances in the methods by which proteins and proteomes are analyzed. Improved fractionation techniques, combined with advances in mass spectrometry, have decreased concerns of sample complexity, and directed more focus towards high-throughput techniques.
Verwandter Inhalt
Standardize Your Research. We offer both the Universal Proteomics Standard and the Proteomics Dynamic range Standard as complex, well-defined, well characterized reference standards for mass spectrometry.
Unser Team von Wissenschaftlern verfügt über Erfahrung in allen Forschungsbereichen einschließlich Life Science, Materialwissenschaften, chemischer Synthese, Chromatographie, Analytik und vielen mehr..
Setzen Sie sich mit dem technischen Dienst in Verbindung.