
B8299
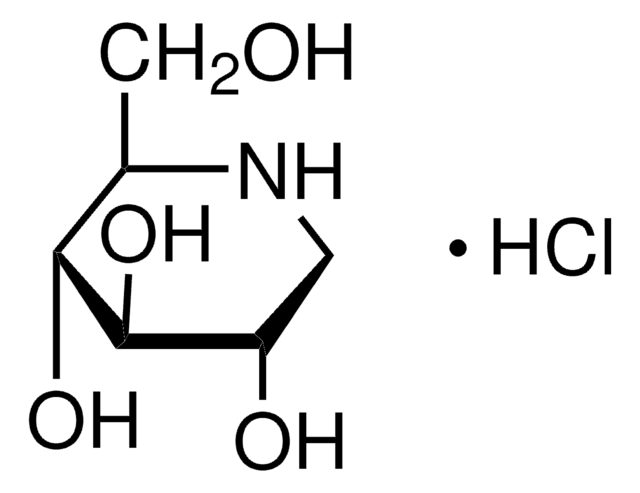
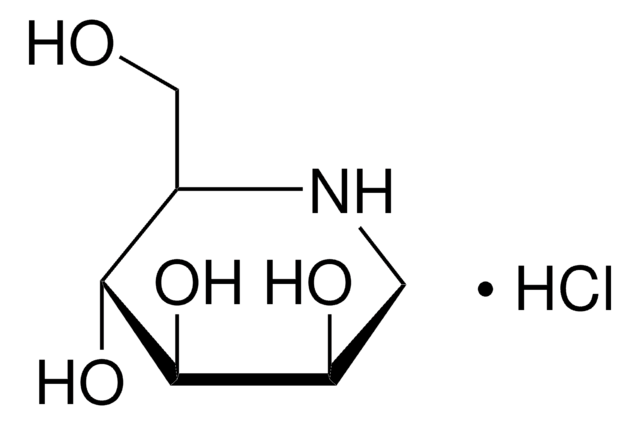
N-Butyldeoxynojirimycin
film (dried in situ), ≥98% (TLC)
Synonym(s):
Miglustat, NB-DNJ
Sign Into View Organizational & Contract Pricing
All Photos(1)
About This Item
Empirical Formula (Hill Notation):
C10H21NO4
CAS Number:
Molecular Weight:
219.28
MDL number:
UNSPSC Code:
12352200
PubChem Substance ID:
NACRES:
NA.32
Recommended Products


Product Name
N-Butyldeoxynojirimycin, film (dried in situ)
Assay
≥98% (TLC)
Quality Level
form
film (dried in situ)
solubility
water: 9.80-10.20 mg/mL, clear, colorless
storage temp.
2-8°C
SMILES string
CCCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO
InChI
1S/C10H21NO4/c1-2-3-4-11-5-8(13)10(15)9(14)7(11)6-12/h7-10,12-15H,2-6H2,1H3/t7-,8+,9-,10-/m1/s1
InChI key
UQRORFVVSGFNRO-UTINFBMNSA-N
Gene Information
human ... UGCG(7357)
Related Categories
General description
N-Butyldeoxynojirimycin is an alkylated product of imino sugar deoxynojirimycin.
Application
N-Butyldeoxynojirimycin has been used:
- in the inhibition of glycolipid synthesis in neuroblastoma cells
- in the inhibition the ceramide-specific glycosyltransferase in hepatocytes
- in the inhibition of β-glucosidase (GBA2) using fluorescence- activity assay in human embryonic kidney (HEK293) cells.
Biochem/physiol Actions
α-glucosidase Inhibitor
N-Butyldeoxynojirimycin is an inhibitor of glucosyltransferase and α-glucosidases. N-Butyldeoxynojirimycin, also known as misglustat, reduces glycolipid levels by substrate reduction therapy (SRT) and is effectively used for the treatment of glycosphingolipid lysosomal storage disorder, Gaucher disease.
Storage Class Code
11 - Combustible Solids
WGK
WGK 3
Flash Point(F)
Not applicable
Flash Point(C)
Not applicable
Personal Protective Equipment
dust mask type N95 (US), Eyeshields, Gloves
Choose from one of the most recent versions:
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Customers Also Viewed
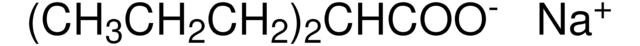
Beom Hee Lee et al.
Medicine, 96(45), e8492-e8492 (2017-11-16)
Gaucher disease (GD) is caused by a deficiency in the lysosomal enzyme glucocerebrosidase. Enzyme replacement therapy (ERT) is recommended for clinical improvement. The efficacy and safety of a new imiglucerase, Abcertin, were assessed in 7 Egyptian patients with treatment-naïve type
Frances M Platt et al.
Nature reviews. Disease primers, 4(1), 27-27 (2018-10-03)
Lysosomal storage diseases (LSDs) are a group of over 70 diseases that are characterized by lysosomal dysfunction, most of which are inherited as autosomal recessive traits. These disorders are individually rare but collectively affect 1 in 5,000 live births. LSDs
Arunabha Ghosh et al.
Orphanet journal of rare diseases, 12(1), 117-117 (2017-06-28)
Mucopolysaccharidosis type III is a progressive, neurodegenerative lysosomal storage disorder for which there is currently no effective therapy. Though numerous potential therapies are in development, there are several challenges to conducting clinical research in this area. We seek to make
Cytoskeletal inhibitors, anti-adhesion molecule antibodies, and lectins inhibit hepatocyte spheroid formation
Nakamura M, et al.
Acta Medica Okayama, 56(1), 43-50 (2002)
The role of the iminosugar N-butyldeoxynojirimycin (miglustat) in the management of type I (non-neuronopathic) Gaucher disease: a position statement
Cox TM, et al.
Journal of inherited metabolic disease, 26(6), 513-526 (2003)
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service