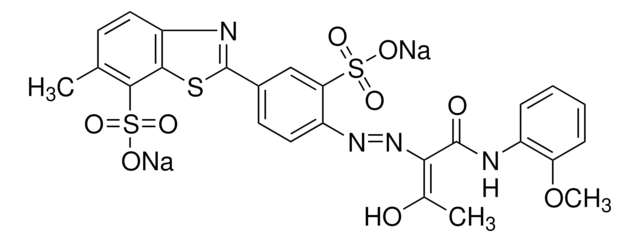
General description
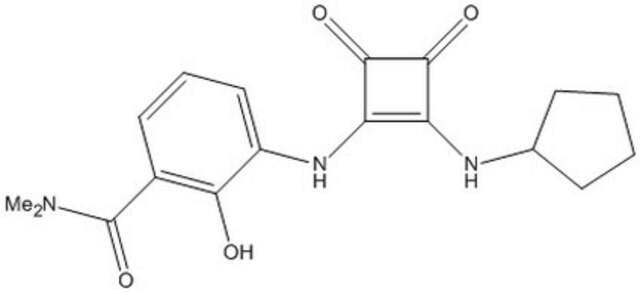
A cell-permeable orally available (plasma Cmax/AUC/dose = 17.0 µM/31.5 µM h-10-24 h/25 mg kg-1/mouse; 5.0 µM/23.0 µM h-10-48 h/10 mg kg-1/rat; 0.9 µM/4.0 µM h-10-48 h/3 mg kg-1/Cynomolgus monkey) cyclobutenedione compound that targets CXCR2 intracellular allosteric site via high affinity interaction (Kd = 49 pM/human, 80 pM/Cynomolgus monkey, 200 pM/ mouse & rat) in a reversible and slowly dissociating manner (k2 = 0.00058 min-1; t1/2 ≈ 22 h; human CXCR2), while exhibiting much reduced affinity toward CXCR1 (Kd = 3.9 nM/human & 41 nM/Cynomolgus monkey) due to a much faster dissociation rate (k2 = 0.141 min-1; t1/2 ≈ 5 min; human CXCR1) and displaying no significant efficacy toward a large panel of GPCRs, enzymes, and ion channels (IC50 >10 µM), including CXCR3 & CCR5. Likewise, Sch527123 is shown exhibit much higher potency against CXCR2- than CXCR1-mediated chemotaxis of Ba/F3 transfectants (IC50/ligand/CXCR2 species = 2.8 nM/3 nM hCXCL8/monkey, 3.4 nM/3 nM rCINC-3/rat, 5.6 nM/mCXCR2/3 nM mMIP-2, <<3 nM/0.1 to 60 nM hCXCL8/human; % inhibition/[Sch527123]/ligand/CXCR1 species = 35%/300 nM/3 nM hCXCL8/ human & 50%/9.667 µM/3 nM hCXCL8/monkey) without affecting C5a- or fMLP-induced human neutrophil chemotaxis. Oral administration is shown to effectively reduce pulmonary neutrophilia in vivo among LPS- or V2O5-treated mice & rats (ED50 1.3 to 1.8 mg/kg), as well as among Cynomolgus monkeys receiving repeat bronchoscopy & lung lavage (ED50 0.3 mg/kg). Also reported to inhibit hCXCL8-dependent human melanoma A375SM chemotaxis & invasion (79% & 69% inhibition, respectively, with 10 ng/mL = 25 nM Sch527123; overnight at 37 °C;10 ng/mL hCXCL8) and effectively suppress A375SM proliferation both in cultures in vitro (31% inhibition by MTT assay post 72 h treatment with 1 µg/mL = 2.5 µM Sch527123) and in mice in vivo (79% inhibition on D50 post cancer cells injection; 100 mg/kg/d p.o. D1 through D22) with concomitant increased apoptotic cells (203% of control) and decreased microvessel density in tumor tissues (55% of control).
A cell-permeable, orally active, non-toxic, cyclobutenedione compound with anti-inflammatory properties. Acts as a specific, high-affinity, and potent allosteric antagonist of CXCR2 (IC50 = 2.6 nM for human; Kd = 49, 200, and 80 pM for human, rat, and cynomolgus, respectively). Also exhibits high potency against CXCR1 (IC50 = 36 nM for human, Kd = 3.9 and 41 nM for human and cynomolgus, respectively). Displays excellent selectivity over CXCR3, CCR5, and a large panel of GPCRs, enzymes and ion channels (~ 10 µM). Potently inhibits CXCL1- and CXCL8-induced chemotaxis in human neutrophils (hPMN; IC50<1 nM and 16 nM, respectively). Shown to suppress pulmonary neutrophilia (ED50 = 1.3 mg/kg), goblet cell hyperplasia (ED50 = 700 mg/kg), and increase bronchoalveolar lavage (BAL) mucin content (ED50 ≤ 1 mg/kg) in rats subjected to vanadium pentoxide induced pulmonary inflammation. Inhibits melanoma cell proliferation (~ 1 mg/ml) by suppressing the phosphorylation of ERK1/2. Diminishes the invasive potential of melanoma cells and blocks angiogenesis in mice (100 mg/kg, p.o., q.d for 21 days). Exhibits desirable pharmacokinetic properties.
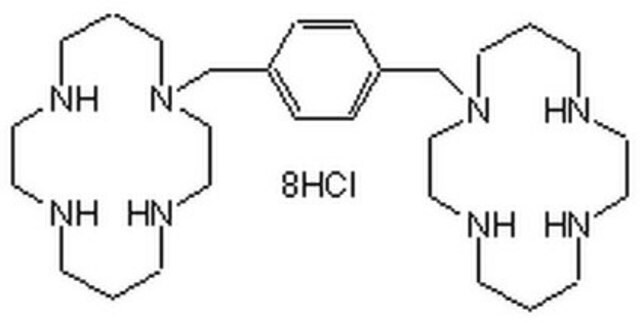
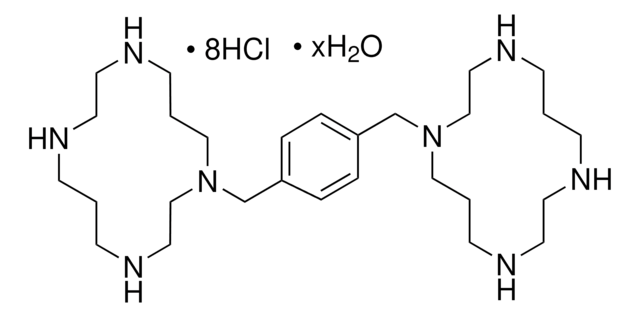
Please note that the molecular weight for this compound is batch-specific due to variable water content.
Reconstitution
Following reconstitution, aliquot and freeze (-20°C). Stock solutions are stable for up to 6 months at -20°C.
Use only fresh DMSO for reconstitution.
Other Notes
Seiberling, M., et al. 2013. Int. Immunopharmacol.17, 178.
Salchow, K., et al. 2010. Br. J. Pharmacol.159, 1429.
Singh, S., et al. 2009. Clin. Cancer Res.15, 2380.
Chapman, R.W., et al. 2007. J. Pharmacol. Exp. Ther.322, 486.
Gonsiorek, W., et al. 2007. J. Pharmacol. Exp. Ther.322, 477.
Dwyer, M.P., et al. 2006. J. Med. Chem.49, 7603.